科研进展
万物皆可Dock:八面钌笼拓展药物化学新空间

英文原题:Structure-based Identification of Organoruthenium Compounds as Nanomolar Antagonists of Cannabinoid Receptors
通讯作者:Jianhui Huang, Niu Huang
作者:Qing Wang, Xuegang Fu, Yuting Yan, Tao Liu, Yuting Xie, Xiaoqing Song, Yu Zhou, Min Xu, Ping Wang, Peng Fu
背景介绍
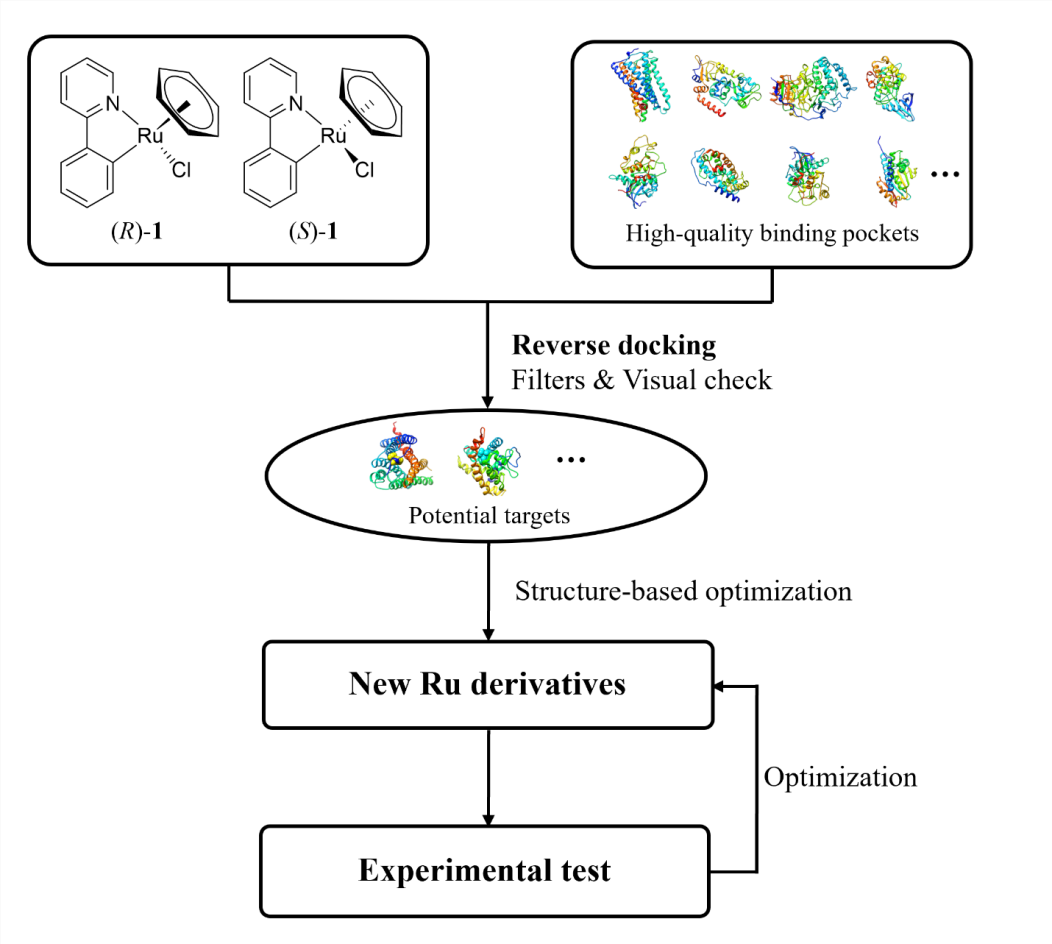
新药研发的失败率居高不下,市场竞争异常激烈,靶标枯竭是重要原因。扩充药物新靶标,一方面需要更深入的生物学研究,另一方面则需要更广阔的化学空间。目前高通量组合化学和DNA编码化合物库等新技术在努力拓展小分子化合物库,但主要还是局限在C、H、O、N、P、S、F、Cl和Br等常见的化学元素中。但纵观元素周期表,70%以上是金属元素,可金属药物却远不及传统的有机药物普遍。是金属元素不能成药吗?不是的,比如大家熟知的铂类药物,自美国生物物理学教授Rosenberg意外发现其具有抑制细菌分裂能力,作为抗癌化疗药物在临床上应用以来,至今仍是多种癌症治疗的一线药物,是TKi或者PD-1抗体等总想逾越的高山。另外,和传统小分子药物与靶标作用方式相比,金属药物与靶标的作用方式可能带来新的药物设计新视角。比如在生物体内,顺铂易发生配体交换,通过与DNA碱基配位发挥抑制肿瘤细胞DNA复制的作用。而且,在三维立体空间上,作为有机化合物中心的碳原子,只能采取线性(sp)、平面(sp2)和四面体(sp3)的几何构型。相比之下,金属原子的几何结构更加多样,可以采取八面体甚至是更复杂的几何构型。利用有机钌化合物的空间立体复杂性,Meggers等人设计了一系列高活性且具有高选择性的激酶抑制剂。这与顺铂的配体交换机制不同,钌原子起到了类似于”八面体碳”的作用,通过与不同的配体螯合形成适应特定激酶结合口袋的独特形状,而非直接与靶标分子作用。受金属配合物立体结构复杂性的启发,该研究利用八面体有机钌化合物,通过反向分子对接等计算方法,以远低于高通量组合化学和DNA编码化合物库的成本就可以去探索分子设计中先前难以触及的化学空间,从而为探索新靶标提供更多可能性。
文章亮点
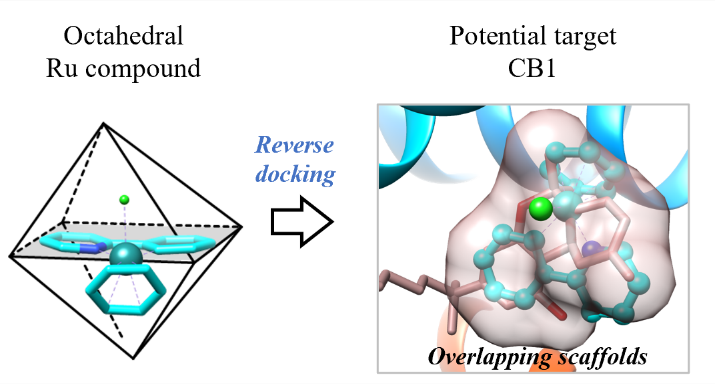
1. 利用反向分子对接方法,发现刚性的含钌对映异构体的对接位置与大麻素受体1(CB1)的共晶配体的骨架在空间上很好的重合,有潜力针对其进行后续的结构优化。
2. 利用分子生长等基于结构的药物设计方法,优化出了一系列对大麻素受体(CB1和CB2)具有纳摩尔结合亲和力的有机钌化合物。其中多个化合物对CB2表现出高选择性,表明可以利用金属配合物的形状复杂性实现靶点特异性。
3. 针对大麻素受体设计的多个有机钌化合物对15-脂氧合酶-2(15-LOX-2)表现出微摩尔级别的抑制活性,说明此类有机钌化合物可以作为一类优势骨架结构结合大且疏水性的蛋白结合口袋。
图文解读
- 反向对接找寻潜在结合靶标
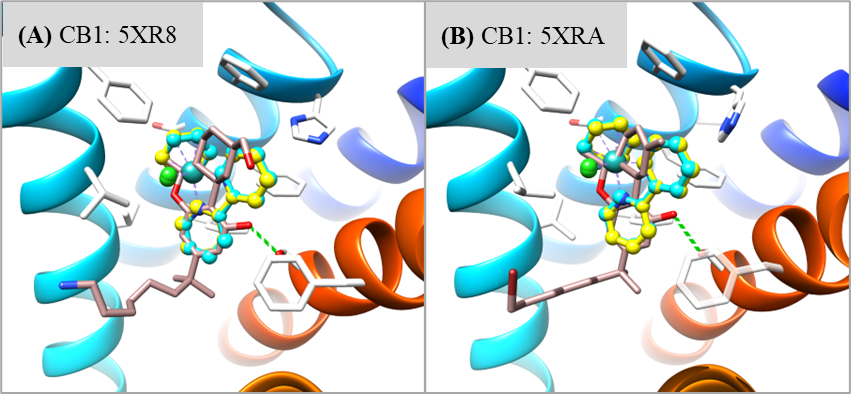
为了加快项目的推进,该研究以一类拥有丰富化学合成经验的半夹心钌(II)配合物作为起点。为了找寻此类含钌骨架的潜在结合口袋,该研究将一对刚性且疏水的含钌异构体((R)-1和(S)-1)反向对接到从PDB数据库中提取的7670个高质量蛋白质结合口袋中(图1)。为方便后续使用骨架跃迁的策略进行结构优化,该研究主要根据对接位置与相应共晶配体的疏水骨架的重叠程度对打分排名靠前的结合口袋进行了过滤。随后经过可视化分析,发现在CB1中,含钌异构体非但与其共晶配体的疏水骨架很好地重叠,还能与结合口袋中的氨基酸残基形成有利的疏水和π-π堆积相互作用(图2)。又考虑到后续结构修饰的可合成性以及生物实验测定的可行性,最终选择大麻素受体进行基于结构的优化设计。
- 针对大麻素受体进行基于结构的优化设计
大麻素受体包含CB1和CB2两个亚型,二者之间的序列同一性约为43%。在竞争性放射配体实验中,基本骨架化合物1和其对伞花烯衍生物2对CB1和CB2均未显示出结合活性(Ki > 40000 nM)。由于这两个化合物只占据结合口袋的一小部分,没有结合活性是意料之中的。为模拟结晶配体的结构,该研究在有机钌骨架的不同位置引入了相似长度的脂肪链。庆幸的是,化合物3、4和5对CB1和CB2均表现出一定结合活性,其中活性最好的化合物5对CB1和CB2的Ki值分别为200和165 nM(图 3)。随后的钙流功能实验表明化合物5对大麻素受体表现出微摩尔级别的拮抗活性(对CB1和CB2的IC50值分别为7439和1700 nM)。随后为降低柔性脂肪链带来的熵的损失,该研究基于化合物5在拮抗剂结合的CB2共晶结构中的对接位置,尝试用苯环取代脂肪链,并利用分子生成的方法在苯环对位生成不同的取代基。在经MM/GBSA重打分以及相互作用分析后,选择并合成了带有正丁基的化合物6和7进行实验验证。与化合物5相比,化合物6和7分别对CB2和CB1的结合活性有3倍左右的提升。化合物7在钙流实验中对CB1(IC50 = 3133 nM)和CB2(IC50 = 2135 nM)也表现出拮抗作用。随后,为了探究化合物的结合特性,该研究合成了一系列化合物7的衍生物,对R2取代基的链长度和吡啶环上不同位置的R1取代基进行了构效关系研究。对于CB1,缩短或延长R2取代基的链长度均会导致结合活性明显降低(6~49倍);在吡啶环的不同位置引入F、Cl、Me或CF3也会导致结合活性有不同程度的降低(2~35倍),其中体积较小的F原子对活性影响最小,体积较大的CF3对活性影响最大。对于CB2,除了去掉正丁基的化合物8 (16倍)、以及CF3取代的化合物16和19(3倍)有明显的活性降低外,其他衍生物对结合活性影响较小(< 2倍)。这些衍生物对CB1和CB2的结合活性影响的程度不同,使得一些化合物对CB2有较高的选择性,尤其是吡啶环第4位的Cl(化合物14,18倍选择性)和Me取代(化合物15,13倍选择性)。由于CB1和CB2拮抗剂结合口袋的大小和形状存在差异,这些化合对CB1和CB2的采用了不同的预测结合模式。但总体而言,化合物对CB1和CB2的预测结合模式与观察到的构效关系保持一致(详情见原文)。
- 结合大且疏水的结合口袋
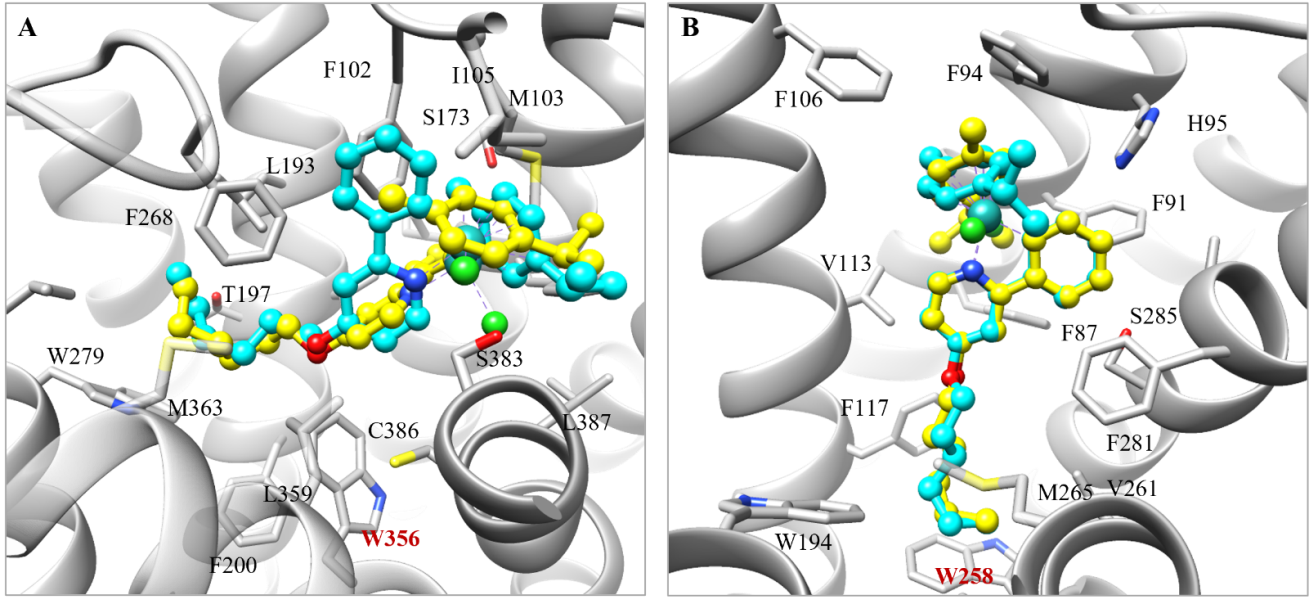
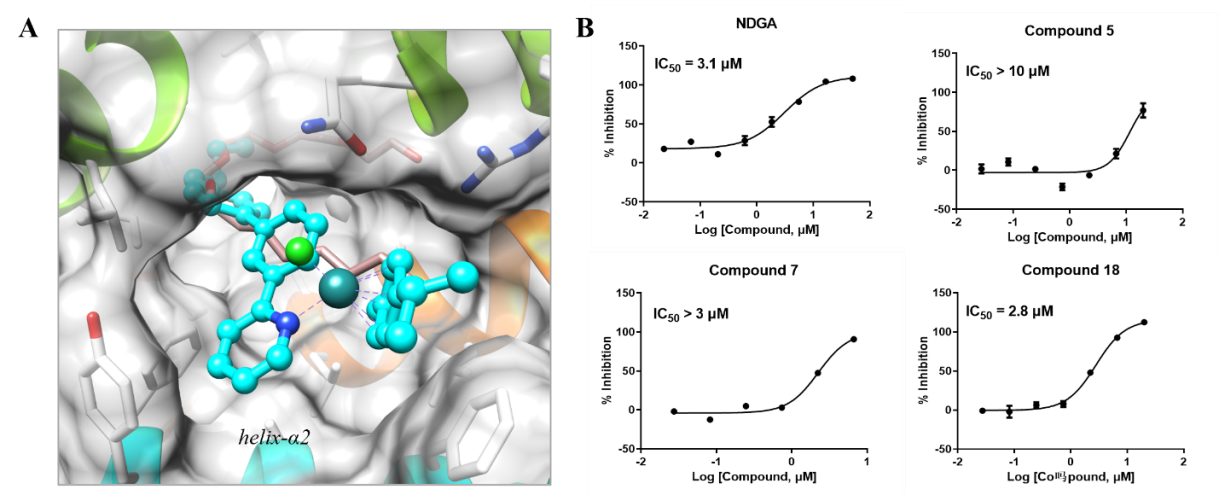
考虑到此类金属有机钌化合物是疏水的,研究者猜想它们很有可能结合在其他大且疏水性的结合口袋中。因此,研究者将化合物7反向对接到以一定标准筛选出的155个大且疏水性的结合口袋中。类似地,根据与共结晶配体的重叠程度,将对接位置进行了筛选。由于化合物7的含钌骨架很好地契合在15-LOX-2的结合口袋中,并且其脂肪链与共结晶配体的朝向类似,该研究最终挑选15-LOX-2进行实验验证(图4A)。在进行单点筛选后,剂量-反应实验结果表明化合物5、7和18均对15-LOX-2表现出微摩尔级别的抑制活性(图4B)。
另外,为了排除此类化合物的结合活性是由胶体聚集导致的非特异性结合造成的,研究者筛选了化合物7对多个靶点的结合活性,包括与大麻素受体具有一定序列相似性的3个GPCR蛋白(LPA1、 MCR4和D1R)、以及3个实验室内部可用的酶(5-LOX、SHP2和COMT)。结果表明,化合物对这些靶点均没有结合活性,说明此类化合物对大麻素受体和15-LOX-2的结合活性不是非特异性结合造成的。
总结与展望
受金属配合物立体结构复杂性的启发,研究者尝试用八面体有机钌化合物来扩展常见有机化合物的化学空间。通过反向分子对接等基于结构的药物设计手段,该研究成功地将一个刚性的八面体有机钌骨架优化为一系列对大麻素受体具有纳摩尔级别结合亲和力的含钌化合物。其中多个化合物对CB2表现出高选择性,表明可以利用金属配合物的形状复杂性实现靶点特异性。同时,其中多个化合物对15-LOX-2表现出微摩尔抑制活性,说明此类有机钌化合物可以作为一类优势骨架结构结合到大且疏水性的蛋白结合口袋中。综上,该研究至少在分子和细胞水平上展示了金属配合物在药物开发中的潜力。值得一提的是,该研究的重点并不是开发CB1或CB2拮抗剂,而是提供一种用于探索金属配合物化学空间的基于结构的药物设计策略。利用这一策略,可以尝试CSD数据库中不同类型的金属有机化合物,通过在药物设计中引入新元素,拓展新的化学空间,从而促进更多新靶点的探索。


